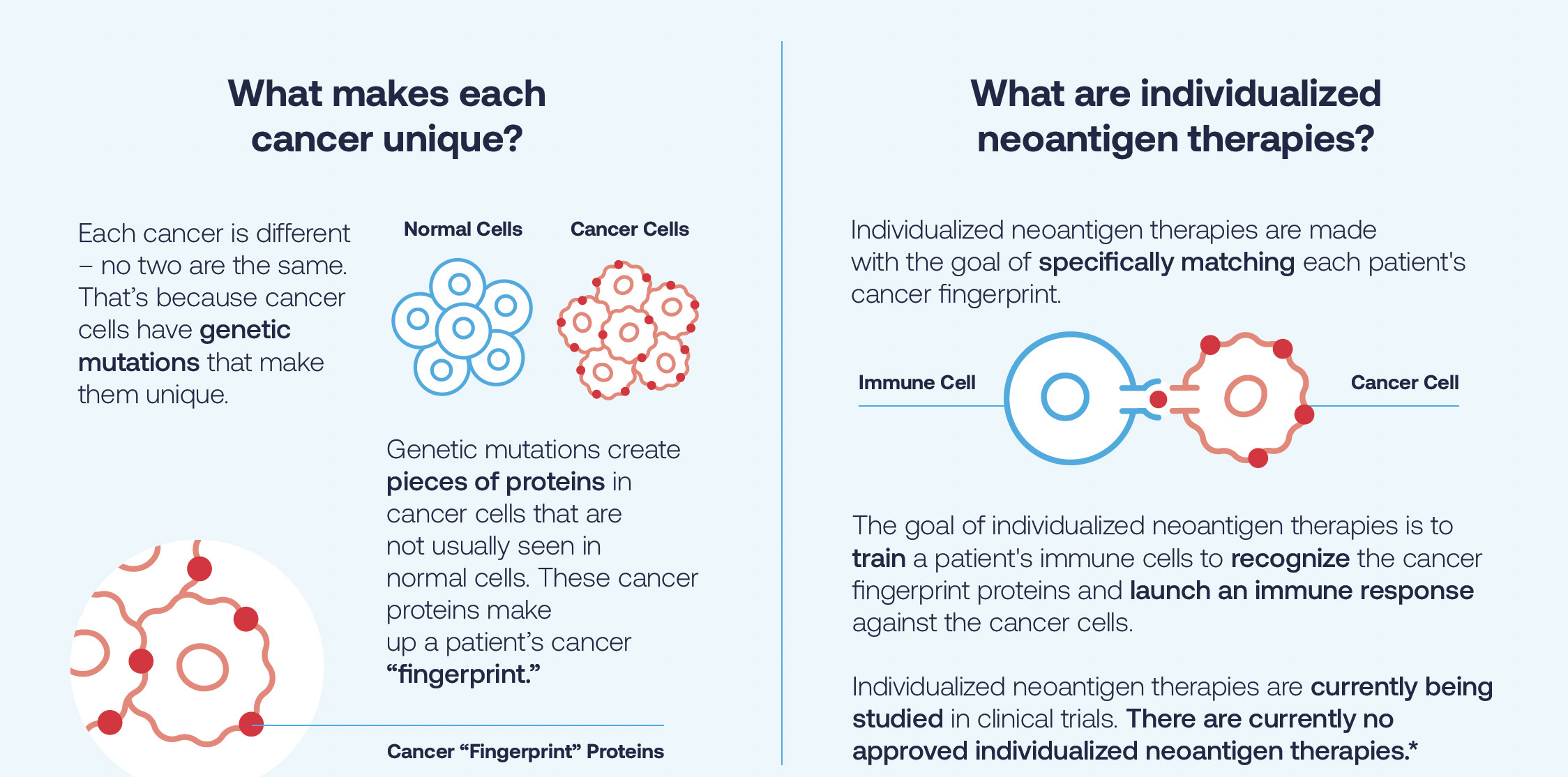
Il cancro di ogni persona è diverso perché le cellule tumorali hanno mutazioni genetiche che le rendono uniche, il che può rendere difficile il trattamento del cancro. Ecco perché noi, in collaborazione con Merck, stiamo ricercando farmaci mRNA progettati specificamente per un singolo paziente. Le terapie neoantigeniche individualizzate (INT) sono un approccio innovativo che adatta il trattamento al singolo tumore di ciascun paziente.
Questa settimana partecipiamo all’American Society for Clinical Oncology (ASCO) Incontro annuale, dove la sopravvivenza libera da metastasi a distanza (DMFS) risulta dallo studio di Fase 2b della nostra terapia neoantigenica individualizzata mRNA sperimentale, mRNA-4157 (V940), in combinazione con KEYTRUDA® (pembrolizumab), la terapia anti-PD-1 di Merck, per il verrà presentato il trattamento adiuvante di pazienti con melanoma in stadio III/IV ad alto rischio dopo resezione completa. L’ASCO Annual Meeting è la più grande conferenza mondiale sull’oncologia e siamo onorati di condividere questi nuovi dati con la comunità oncologica globale.
Siamo incredibilmente grati ai partecipanti alle nostre sperimentazioni cliniche e ai ricercatori e al personale delle sperimentazioni cliniche che stanno aiutando a far progredire questa importante ricerca sul cancro.
Una medicina per un paziente: individualizzare la ricerca sul cancro
Anche dopo 50 anni di studi clinici, gli approcci basati sui vaccini antitumorali hanno avuto scarso successo, poiché nessun regime contenente vaccini antitumorali ha mostrato benefici in termini di sopravvivenza libera da recidiva rispetto allo standard di cura tradizionale.¹
Gli sviluppi nel campo dell’immunoterapia hanno trasformato la nostra comprensione del ruolo del sistema immunitario nella lotta contro alcuni tipi di cancro. Basandoci sugli sviluppi della comprensione immunologica e della biologia del cancro, stiamo ricercando e sviluppando un approccio terapeutico individualizzato specifico per il tumore di ciascun paziente. Moderna, in collaborazione con Merck, sta esplorando gli INT come opzione terapeutica progettata per allenare e attivare potenzialmente il sistema immunitario per aumentare la sua capacità di combattere il cancro. Con questo lavoro, speriamo di far avanzare ulteriormente il campo dell’immuno-oncologia.
Le cellule tumorali esprimono proteine ??anormali, o neoantigeni, che di solito non si vedono nelle cellule normali. Queste proteine ????del cancro costituiscono l’impronta digitale del cancro di un paziente. Le terapie neoantigeniche individualizzate sono progettate con l’obiettivo di abbinare in modo specifico l’impronta digitale unica del cancro di un paziente. L’obiettivo delle terapie neoantigeniche individualizzate è addestrare le cellule immunitarie di un paziente a riconoscere le proteine ??dell’impronta digitale del cancro e lanciare una risposta immunitaria contro le cellule tumorali.
Le terapie neoantigeniche individualizzate sono attualmente oggetto di studi clinici. Ad oggi, non esistono attualmente terapie neoantigene individualizzate approvate dalla FDA.
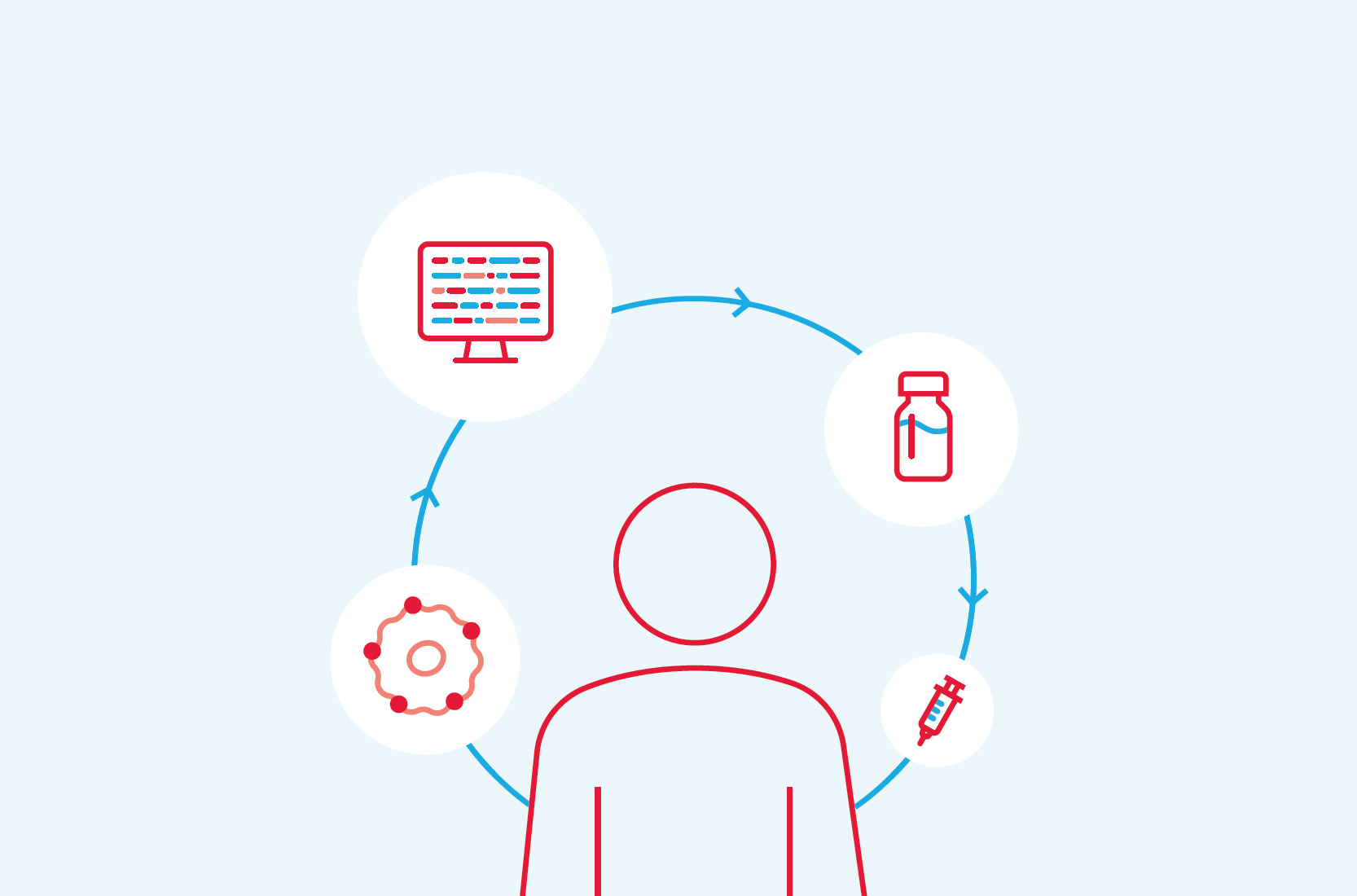
Come vengono realizzate le terapie neoantigeniche individualizzate con mRNA sperimentale?
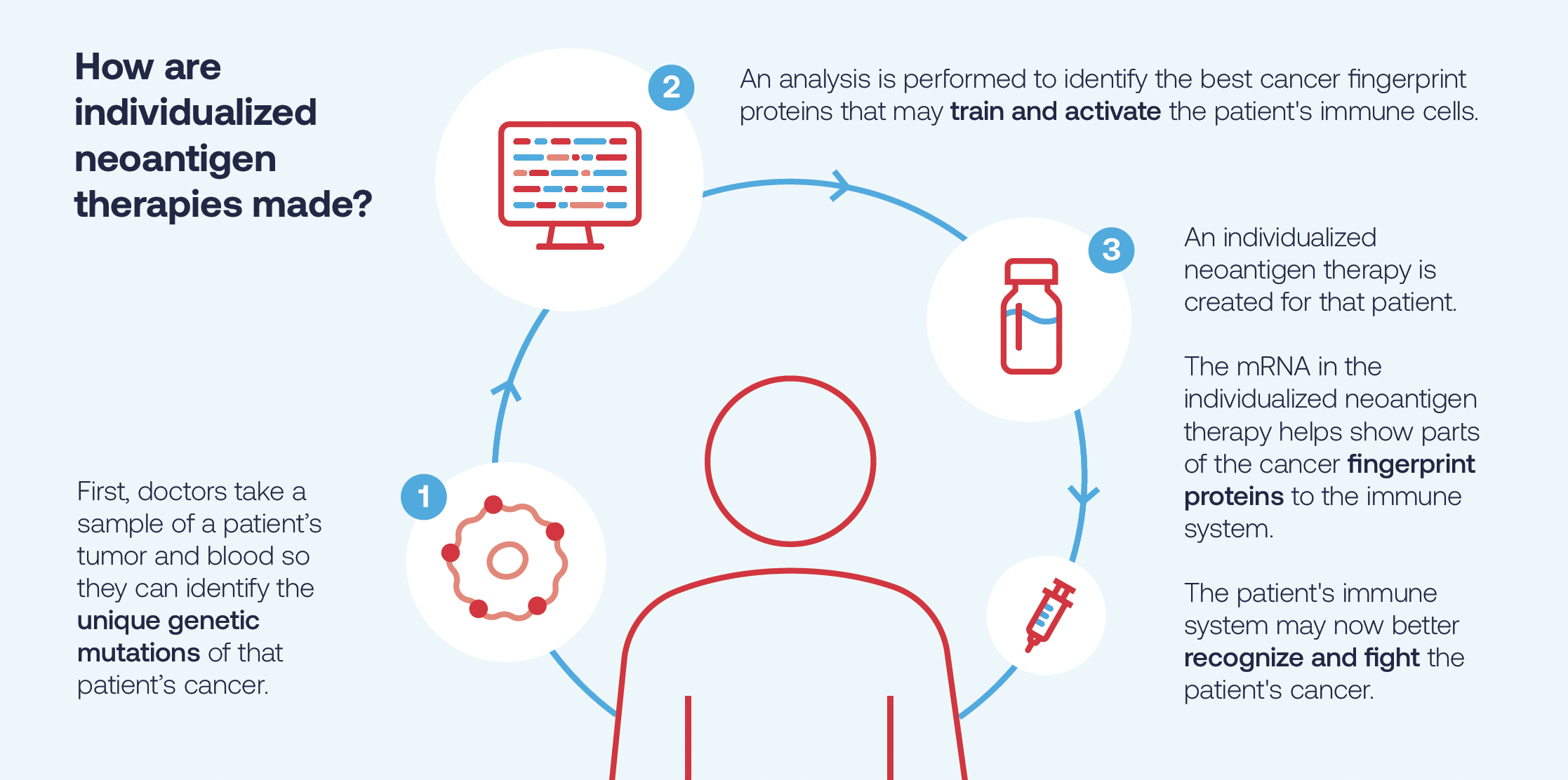
Il processo per realizzare la nostra terapia neoantigenica individualizzata inizia in clinica, dove un partecipante allo studio viene prelevato il sangue e sottoposto a biopsia del tumore. Il sequenziamento viene quindi eseguito in un laboratorio per confrontare i dati del campione bioptico del tumore e le cellule del sangue sane del partecipante allo studio, che non sono mutate, per identificare le mutazioni genetiche uniche del cancro di quell’individuo.
Un algoritmo proprietario sviluppato in collaborazione con Merck esamina quindi queste mutazioni e prevede fino a 34 di quelle mutazioni che si ritiene aiutino il sistema immunitario del partecipante allo studio a riconoscere meglio le cellule tumorali e a lanciare un attacco. Successivamente, viene creata la terapia neoantigenica individualizzata sperimentale per dare istruzioni alle cellule per produrre la proteina cancerosa “impronta digitale”, con l’obiettivo di aiutare il corpo a riconoscere e attaccare il cancro unico di quel partecipante allo studio.
Dopo la fase di progettazione, queste informazioni vengono quindi inviate al nostro impianto di produzione a Norwood, MA, dove viene prodotta la terapia neoantigenica individualizzata. La terapia neoantigenica individualizzata viene quindi inserita in una fiala e rispedita al sito della sperimentazione clinica dove un operatore sanitario la somministra al paziente dello studio tramite iniezione intramuscolare.
Dati presentati alla riunione annuale dell’ASCO
Oggi presentiamo i risultati della sopravvivenza libera da metastasi a distanza (DMFS) dello studio di fase 2b randomizzato KEYNOTE-942/mRNA-4157-P201. Nella popolazione complessiva intent-to-treat, il trattamento adiuvante con mRNA-4157 (V940) in combinazione con KEYTRUDA ha dimostrato un miglioramento statisticamente significativo e clinicamente significativo della DMFS, un endpoint secondario chiave dello studio, rispetto al solo KEYTRUDA e ha ridotto il rischio di sviluppare metastasi a distanza o morte del 65% (HR = 0,347 [95% CI, 0,145-0,828]); valore p unilaterale = 0,0063). L’endpoint secondario della DMFS, definito come il tempo dalla prima dose di KEYTRUDA fino alla data della prima recidiva a distanza o decesso per qualsiasi causa, è stato pre-specificato per i test statistici dopo l’endpoint primario positivo della sopravvivenza libera da recidiva (RFS).
Gli eventi avversi riportati con mRNA-4157 (V940) in KEYNOTE-942 erano coerenti con quelli precedentemente osservati in uno studio clinico di fase 1. Il profilo di sicurezza di KEYTRUDA era coerente con i risultati di studi precedenti. Il numero di pazienti che hanno riportato eventi avversi di Grado ? 3 correlati al trattamento è stato simile tra i bracci (25% vs. 18%, rispettivamente). Gli eventi avversi più comuni di qualsiasi grado attribuiti a mRNA-4157 (V940) o alla combinazione di mRNA-4157 (V940) e KEYTRUDA sono stati affaticamento (60,6%), dolore al sito di iniezione (55,8%) e brividi (50,0%).
All’ASCO, presentiamo anche i dati di un’analisi esplorativa di sottogruppi di KEYNOTE-942/mRNA-4157-P201, valutando la malattia minima residua (MRD) mediante la circolazione del DNA tumorale (ctDNA) come biomarcatore della sopravvivenza libera da recidiva (RFS) in pazienti con melanoma ad alto rischio resecati trattati con mRNA-4157 (V940) in combinazione con KEYTRUDA.
Studio di fase 2b sulla terapia neoantigenica individualizzata con mRNA: sopravvivenza libera da recidiva
Con Merck, stiamo studiando la nostra terapia neoantigenica individualizzata con mRNA somministrata in combinazione con KEYTRUDA rispetto al solo KEYTRUDA. Abbiamo selezionato KEYTRUDA come comparatore nello studio KEYNOTE-942/mRNA-4157-P201 perché è considerato uno standard di cura nel melanoma adiuvante. 107 pazienti hanno ricevuto la combinazione del nostro mRNA-4157 sperimentale (V940) con KEYTRUDA e 50 pazienti sono stati trattati solo con KETRUDA.
Ad aprile, all’incontro annuale dell’American Association for Cancer Research (AACR), abbiamo presentato i dati sull’endpoint primario di sopravvivenza libera da recidiva (RFS) dello studio di fase 2b KEYNOTE-942/mRNA-4157-P201 in pazienti con stadio III/IV melanoma dopo resezione completa. Questi dati hanno mostrato che mRNA-4157 (V940) in combinazione con KEYTRUDA ha dimostrato un miglioramento statisticamente significativo e clinicamente significativo nella RFS rispetto a KEYTRUDA da solo.
Nello specifico, la terapia neoantigenica individualizzata in combinazione con KEYTRUDA ha ridotto il rischio di recidiva o morte del 44% (HR = 0,56 [IC 95%, 0,31-1,017]; valore p unilaterale = 0,0266), rispetto al solo KEYTRUDA.
Gli eventi avversi riportati con mRNA-4157 (V940) in KEYNOTE-942 precedentemente presentati all’AACR erano coerenti con quelli attualmente condivisi all’ASCO.
Sulla base dei dati dello studio di fase 2b, mRNA-4157 (V940) in combinazione con KEYTRUDA per il trattamento adiuvante di pazienti con melanoma ad alto rischio dopo resezione completa ha ottenuto la designazione di terapia innovativa dalla Food and Drug Administration (FDA) statunitense ed è stato approvato dallo schema PRIME (Priority Medicines) dall’Agenzia europea per i medicinali.
Guardando avanti
Questi risultati aggiungono una maggiore comprensione della potenziale promessa di un approccio neoantigenico individualizzato mRNA per affrontare il cancro. Guardando al futuro, in collaborazione con Merck, prevediamo di avviare uno studio di Fase 3 sul melanoma nel 2023 e di espandere rapidamente i nostri sforzi per studiare altri tipi di tumore, incluso il carcinoma polmonare non a piccole cellule. Continueremo a ricercare approcci neoantigenici individualizzati con l’obiettivo di aiutare potenzialmente più pazienti con cancro.
Dichiarazioni lungimiranti di Moderna
Questo post contiene dichiarazioni previsionali ai sensi del Private Securities Litigation Reform Act del 1995, come modificato, incluse dichiarazioni riguardanti: lo sviluppo da parte di Moderna e Merck di una terapia neoantigenica individualizzata (mRNA-4157/V940); la capacità e il potenziale dell’mRNA-4157/V940 di migliorare la sopravvivenza libera da metastasi a distanza (DMFS) in pazienti con melanoma in stadio III/IV ad alto rischio; la capacità e il potenziale dell’mRNA-4157/V940 di migliorare i tassi di sopravvivenza libera da recidiva (RFS) nei pazienti con melanoma in stadio III/IV; il profilo di tollerabilità e sicurezza per mRNA-4157/V940; il potenziale dell’mRNA, incluso l’mRNA-4157, per trattare efficacemente diversi tipi di cancro; prevede di avviare uno studio di fase 3 sul melanoma nel 2023 e di espandersi rapidamente ad altri tipi di tumore, compreso il carcinoma polmonare non a piccole cellule; il potenziale sviluppo di terapie e trattamenti neoantigenici individualizzati la capacità di una terapia neoantigenica individualizzata di innescare una risposta antitumorale su misura specifica per la firma della mutazione tumorale di un paziente; e il potenziale per l’approvazione normativa e la commercializzazione di mRNA-4157/V940 e le implicazioni per la designazione dello schema PRIME dell’EMA per mRNA-4157/V940 e la designazione di terapia rivoluzionaria della FDA per mRNA-4157/V940. In alcuni casi, le dichiarazioni previsionali possono essere identificate da terminologia come “sarà”, “può”, “dovrebbe”, “potrebbe”, “si aspetta”, “intende”, “pianifica”, “mira”, “prevede, ” “crede”, “stima”, “predice”, “potenziale”, “continua”, o il negativo di questi termini o altra terminologia comparabile, sebbene non tutte le dichiarazioni previsionali contengano queste parole. Le dichiarazioni previsionali in questo post non sono né promesse né garanzie, e non dovresti fare eccessivo affidamento su queste dichiarazioni previsionali perché comportano rischi noti e sconosciuti, incertezze e altri fattori, molti dei quali sono al di fuori del controllo di Moderna e che potrebbe far sì che i risultati effettivi differiscano materialmente da quelli espressi o impliciti in queste dichiarazioni previsionali. Questi rischi, incertezze e altri fattori includono, tra gli altri, quei rischi e incertezze descritti sotto il titolo “Fattori di rischio” nella relazione annuale di Moderna sul modulo 10-K per l’anno fiscale terminato il 31 dicembre 2022 depositata presso la US Securities and Exchange Commissione (SEC), e nei successivi documenti depositati da Moderna presso la SEC, disponibili sul sito Web della SEC all’indirizzo http://www.sec.gov. Ad eccezione di quanto richiesto dalla legge, Moderna declina ogni intenzione o responsabilità per l’aggiornamento o la revisione di eventuali dichiarazioni previsionali contenute in questo post in caso di nuove informazioni, sviluppi futuri o altro. Queste dichiarazioni previsionali si basano sulle aspettative attuali di Moderna e parlano solo alla data di questo post.
Informazioni sul melanoma La forma più grave di cancro della pelle, il melanoma è caratterizzato dalla crescita incontrollata delle cellule produttrici di pigmento. I tassi di melanoma sono aumentati negli ultimi decenni, con quasi 325.000 nuovi casi diagnosticati in tutto il mondo nel 2020. Negli Stati Uniti, il cancro della pelle è uno dei tipi più comuni di cancro diagnosticati e il melanoma rappresenta la maggior parte dei tumori della pelle deceduti. Si stima che ci saranno quasi 100.000 nuovi casi di melanoma diagnosticati e quasi 8.000 decessi derivanti dalla malattia negli Stati Uniti nel 2022.
Informazioni sull’iniezione di KEYTRUDA® (pembrolizumab), 100 mg KEYTRUDA è una terapia anti-recettore della morte programmata-1 (PD-1) che agisce aumentando la capacità del sistema immunitario del corpo di aiutare a rilevare e combattere le cellule tumorali. KEYTRUDA è un anticorpo monoclonale umanizzato che blocca l’interazione tra PD-1 e i suoi ligandi, PD-L1 e PD-L2, attivando così i linfociti T che possono interessare sia le cellule tumorali che le cellule sane. Merck ha il più grande programma di ricerca clinica immuno-oncologica del settore. Attualmente ci sono più di 1.600 studi che studiano KEYTRUDA in un’ampia varietà di tumori e impostazioni terapeutiche. Il programma clinico KEYTRUDA cerca di comprendere il ruolo di KEYTRUDA nei tumori e i fattori che possono prevedere la probabilità di un paziente di trarre beneficio dal trattamento con KEYTRUDA, inclusa l’esplorazione di diversi biomarcatori.
Indicazioni selezionate di KEYTRUDA® (pembrolizumab) negli Stati Uniti Melanoma KEYTRUDA è indicato per il trattamento di pazienti con melanoma non resecabile o metastatico. KEYTRUDA è indicato per il trattamento adiuvante di pazienti adulti e pediatrici (dai 12 anni in su) con melanoma in stadio IIB, IIC o III dopo resezione completa.
Carcinoma polmonare non a piccole cellule KEYTRUDA, in combinazione con pemetrexed e chemioterapia a base di platino, è indicato per il trattamento di prima linea di pazienti con carcinoma polmonare non a piccole cellule metastatico non squamoso (NSCLC), senza aberrazioni del tumore genomico di EGFR o ALK. KEYTRUDA, in combinazione con carboplatino e paclitaxel o paclitaxel legato alle proteine, è indicato per il trattamento di prima linea di pazienti con NSCLC squamoso metastatico. KEYTRUDA, come agente singolo, è indicato per il trattamento di prima linea di pazienti con NSCLC che esprimono PD-L1 [punteggio di proporzione tumorale (TPS) ?1%] come determinato da un test approvato dalla FDA, senza tumore genomico di EGFR o ALK aberrazioni, ed è: -stadio III in cui i pazienti non sono candidati alla resezione chirurgica o alla chemioradioterapia definitiva, o -metastatico. KEYTRUDA, in monoterapia, è indicato per il trattamento di pazienti con NSCLC metastatico i cui tumori esprimono PD-L1 (TPS ?1%) come determinato da un test approvato dalla FDA, con progressione della malattia durante o dopo chemioterapia contenente platino. I pazienti con aberrazioni tumorali genomiche EGFR o ALK devono avere una progressione della malattia in terapia approvata dalla FDA per queste aberrazioni prima di ricevere KEYTRUDA.
Carcinoma a cellule squamose della testa e del collo KEYTRUDA, in combinazione con platino e fluorouracile (FU), è indicato per il trattamento di prima linea di pazienti con carcinoma a cellule squamose della testa e del collo (HNSCC) metastatico o non resecabile e ricorrente. KEYTRUDA, in monoterapia, è indicato per il trattamento di prima linea di pazienti con HNSCC metastatico o non resecabile e ricorrente i cui tumori esprimono PD-L1 [Combined Positive Score (CPS) ?1] come determinato da un test approvato dalla FDA. KEYTRUDA, in monoterapia, è indicato per il trattamento di pazienti con HNSCC ricorrente o metastatico con progressione della malattia durante o dopo chemioterapia contenente platino.
Linfoma di Hodgkin classico KEYTRUDA è indicato per il trattamento di pazienti adulti affetti da linfoma di Hodgkin classico (cHL) recidivante o refrattario. KEYTRUDA è indicato per il trattamento di pazienti pediatrici con cHL refrattario o con cHL recidivante dopo 2 o più linee di terapia.
Linfoma primario del mediastino a grandi cellule B KEYTRUDA è indicato per il trattamento di pazienti adulti e pediatrici con linfoma primario del mediastino a grandi cellule B (PMBCL) refrattario o che hanno avuto una recidiva dopo 2 o più linee di terapia precedenti. KEYTRUDA non è raccomandato per il trattamento di pazienti con PMBCL che richiedono una terapia citoriduttiva urgente.
Carcinoma uroteliale KEYTRUDA è indicato per il trattamento di pazienti con carcinoma uroteliale (mUC) localmente avanzato o metastatico: – che non sono eleggibili per alcuna chemioterapia contenente platino, o – che hanno una progressione della malattia durante o dopo la chemioterapia contenente platino o entro 12 mesi di trattamento neoadiuvante o adiuvante con chemioterapia contenente platino.
Carcinoma vescicale non muscolo-invasivo KEYTRUDA è indicato per il trattamento di pazienti affetti da carcinoma vescicale non-muscolo-invasivo (NMIBC) non responsivo, ad alto rischio, con carcinoma in situ con o senza tumori papillari, non responsivi al Bacillus Calmette-Guerin, non idonei per o che hanno scelto di non sottoporsi a cistectomia.
Microsatellite Instability-High o Mismatch Repair Deficient Cancer KEYTRUDA è indicato per il trattamento di pazienti adulti e pediatrici con tumori solidi non resecabili o metastatici ad alta instabilità dei microsatelliti (MSI-H) o mismatch repair deficient (dMMR), come determinato da un test approvato dalla FDA test, che sono progrediti dopo un trattamento precedente e che non hanno opzioni terapeutiche alternative soddisfacenti.
Instabilità dei microsatelliti: riparazione del cancro colorettale carente o mancata corrispondenza KEYTRUDA è indicato per il trattamento di pazienti con carcinoma del colon-retto (CRC) non operabile o metastatico con MSI-H o dMMR, come determinato da un test approvato dalla FDA.
Cancro gastrico KEYTRUDA, in combinazione con trastuzumab, chemioterapia contenente fluoropirimidine e platino, è indicato per il trattamento di prima linea di pazienti con adenocarcinoma gastrico o della giunzione gastroesofagea (GEJ) localmente avanzato non resecabile o metastatico HER2-positivo. Questa indicazione è approvata con approvazione accelerata in base al tasso di risposta del tumore e alla durata della risposta. L’approvazione continua per questa indicazione può essere subordinata alla verifica e alla descrizione del beneficio clinico negli studi di conferma.
Cancro esofageo KEYTRUDA è indicato per il trattamento di pazienti con carcinoma della giunzione gastroesofagea o esofagea (GEJ) localmente avanzato o metastatico (tumori con epicentro da 1 a 5 centimetri al di sopra del GEJ) non suscettibili di resezione chirurgica o chemioradioterapia definitiva: – in combinazione con platino – e chemioterapia a base di fluoropirimidine, o – come agente singolo dopo una o più linee precedenti di terapia sistemica per pazienti con tumori a istologia a cellule squamose che esprimono PD-L1 (CPS ?10) come determinato da un test approvato dalla FDA.
Cancro cervicale KEYTRUDA, in combinazione con chemioterapia, con o senza bevacizumab, è indicato per il trattamento di pazienti con carcinoma cervicale persistente, ricorrente o metastatico i cui tumori esprimono PD-L1 (CPS ?1) come determinato da un test approvato dalla FDA. KEYTRUDA, in monoterapia, è indicato per il trattamento di pazienti con carcinoma della cervice uterina ricorrente o metastatico con progressione della malattia durante o dopo la chemioterapia i cui tumori esprimono PD-L1 (CPS ?1) come determinato da un test approvato dalla FDA.
Carcinoma epatocellulare KEYTRUDA è indicato per il trattamento di pazienti con carcinoma epatocellulare (HCC) che sono stati precedentemente trattati con sorafenib. Questa indicazione è approvata con approvazione accelerata in base al tasso di risposta del tumore e alla durata della risposta. L’approvazione continua per questa indicazione può essere subordinata alla verifica e alla descrizione del beneficio clinico negli studi di conferma.
Carcinoma a cellule di Merkel KEYTRUDA è indicato per il trattamento di pazienti adulti e pediatrici affetti da carcinoma a cellule di Merkel (MCC) ricorrente, localmente avanzato o metastatico. Questa indicazione è approvata con approvazione accelerata in base al tasso di risposta del tumore e alla durata della risposta. L’approvazione continua per questa indicazione può essere subordinata alla verifica e alla descrizione del beneficio clinico negli studi di conferma.
Carcinoma a cellule renali KEYTRUDA, in combinazione con axitinib, è indicato per il trattamento di prima linea di pazienti adulti con carcinoma a cellule renali (RCC) avanzato. KEYTRUDA è indicato per il trattamento adiuvante di pazienti con RCC a rischio di recidiva intermedio-alto o alto dopo nefrectomia o dopo nefrectomia e resezione di lesioni metastatiche.
Carcinoma endometriale KEYTRUDA, come agente singolo, è indicato per il trattamento di pazienti con carcinoma endometriale avanzato che è MSI-H o dMMR, come determinato da un test approvato dalla FDA, che hanno una progressione della malattia dopo una precedente terapia sistemica in qualsiasi contesto e non sono candidati per chirurgia curativa o radioterapia.
Tumor Mutational Burden-High Cancer KEYTRUDA è indicato per il trattamento di pazienti adulti e pediatrici con tumori solidi non resecabili o metastatici ad alto carico mutazionale tumorale (TMB-H) [?10 mutazioni/megabase], come determinato da un test approvato dalla FDA, che sono progrediti dopo un trattamento precedente e che non hanno opzioni terapeutiche alternative soddisfacenti. Questa indicazione è approvata con approvazione accelerata in base al tasso di risposta del tumore e alla durata della risposta. L’approvazione continua per questa indicazione può essere subordinata alla verifica e alla descrizione del beneficio clinico negli studi di conferma. La sicurezza e l’efficacia di KEYTRUDA nei pazienti pediatrici con tumori del sistema nervoso centrale TMB-H non sono state stabilite.
Carcinoma cutaneo a cellule squamose KEYTRUDA è indicato per il trattamento di pazienti con carcinoma cutaneo a cellule squamose (cSCC) ricorrente o metastatico o con cSCC localmente avanzato non curabile mediante chirurgia o radioterapia.
Carcinoma mammario triplo negativo KEYTRUDA è indicato per il trattamento di pazienti con carcinoma mammario triplo negativo (TNBC) in stadio iniziale ad alto rischio in combinazione con chemioterapia come trattamento neoadiuvante, e poi proseguito come singolo agente come trattamento adiuvante dopo l’intervento chirurgico. KEYTRUDA, in combinazione con la chemioterapia, è indicato per il trattamento di pazienti con TNBC non resecabile o metastatico localmente ricorrente i cui tumori esprimono PD-L1 (CPS ?10) come determinato da un test approvato dalla FDA. Importanti informazioni sulla sicurezza selezionate per KEYTRUDA Reazioni avverse immuno-mediate gravi e fatali KEYTRUDA è un anticorpo monoclonale che appartiene a una classe di farmaci che si legano al PD-1 o al PD-L1, bloccando la via PD-1/PD-L1, rimuovendo così l’inibizione del risposta immunitaria, rompendo potenzialmente la tolleranza periferica e inducendo reazioni avverse immuno-mediate. Le reazioni avverse immuno-mediate, che possono essere gravi o fatali, possono verificarsi in qualsiasi sistema di organi o tessuti, possono interessare più di un sistema corporeo contemporaneamente e possono verificarsi in qualsiasi momento dopo l’inizio del trattamento o dopo l’interruzione del trattamento. Le reazioni avverse immuno-mediate importanti qui elencate potrebbero non includere tutte le possibili reazioni avverse immuno-mediate gravi e fatali.
Monitorare attentamente i pazienti per sintomi e segni che possono essere manifestazioni cliniche di reazioni avverse immuno-mediate sottostanti. L’identificazione e la gestione precoci sono essenziali per garantire un uso sicuro dei trattamenti anti-PD-1/PD-L1. Valutare gli enzimi epatici, la creatinina e la funzione tiroidea al basale e periodicamente durante il trattamento. Per i pazienti con TNBC trattati con KEYTRUDA in ambito neoadiuvante, monitorare il cortisolo ematico al basale, prima dell’intervento chirurgico e come clinicamente indicato. In caso di sospette reazioni avverse immuno-mediate, avviare una valutazione appropriata per escludere eziologie alternative, inclusa l’infezione. Istituire tempestivamente la gestione medica, compresa la consulenza specialistica, se del caso.
Sospendere o interrompere definitivamente KEYTRUDA a seconda della gravità della reazione avversa immuno-mediata. In generale, se KEYTRUDA richiede l’interruzione o l’interruzione, somministrare una terapia con corticosteroidi sistemici (da 1 a 2 mg/kg/die di prednisone o equivalente) fino al miglioramento al Grado 1 o inferiore. Dopo il miglioramento al Grado 1 o inferiore, iniziare la riduzione graduale dei corticosteroidi e continuare a diminuire per almeno 1 mese. Considerare la somministrazione di altri immunosoppressori sistemici nei pazienti le cui reazioni avverse non sono controllate con la terapia con corticosteroidi.
Polmonite immuno-mediata KEYTRUDA può causare polmonite immuno-mediata. L’incidenza è maggiore nei pazienti che hanno ricevuto una precedente radioterapia toracica. Polmonite immuno-mediata si è verificata nel 3,4% (94/2799) dei pazienti trattati con KEYTRUDA, comprese reazioni fatali (0,1%), di grado 4 (0,3%), di grado 3 (0,9%) e di grado 2 (1,3%). I corticosteroidi sistemici sono stati richiesti nel 67% (63/94) dei pazienti. La polmonite ha portato all’interruzione permanente di KEYTRUDA nell’1,3% (36) e alla sospensione nello 0,9% (26) dei pazienti. Tutti i pazienti che sono stati sospesi hanno ripreso KEYTRUDA dopo il miglioramento dei sintomi; di questi, il 23% ha avuto recidiva. La polmonite si è risolta nel 59% dei 94 pazienti.
La polmonite si è verificata nell’8% (31/389) dei pazienti adulti con cHL trattati con KEYTRUDA in monoterapia, compresi i Gradi 3-4 nel 2,3% dei pazienti. I pazienti hanno ricevuto corticosteroidi ad alte dosi per una durata mediana di 10 giorni (intervallo: da 2 giorni a 53 mesi). I tassi di polmonite erano simili nei pazienti con e senza precedente radioterapia toracica. La polmonite ha portato all’interruzione di KEYTRUDA nel 5,4% (21) dei pazienti. Dei pazienti che hanno sviluppato polmonite, il 42% ha interrotto KEYTRUDA, il 68% ha interrotto KEYTRUDA e il 77% ha avuto una risoluzione.
Colite immunomediata KEYTRUDA può causare colite immuno-mediata, che può presentarsi con diarrea. Infezione/riattivazione da citomegalovirus è stata segnalata in pazienti con colite immuno-mediata refrattaria ai corticosteroidi. Nei casi di colite refrattaria ai corticosteroidi, prendere in considerazione la ripetizione del workup infettivo per escludere eziologie alternative. Colite immuno-mediata si è verificata nell’1,7% (48/2799) dei pazienti trattati con KEYTRUDA, comprese reazioni di Grado 4 (<0,1%), Grado 3 (1,1%) e Grado 2 (0,4%). I corticosteroidi sistemici sono stati richiesti nel 69% (33/48); ulteriore terapia immunosoppressiva è stata richiesta nel 4,2% dei pazienti. La colite ha portato all’interruzione permanente di KEYTRUDA nello 0,5% (15) e alla sospensione nello 0,5% (13) dei pazienti. Tutti i pazienti che sono stati sospesi hanno ripreso KEYTRUDA dopo il miglioramento dei sintomi; di questi, il 23% ha avuto recidiva.
Epatotossicità ed epatite immuno-mediata KEYTRUDA come agente singolo KEYTRUDA può causare epatite immuno-mediata. L’epatite immuno-mediata si è verificata nello 0,7% (19/2799) dei pazienti trattati con KEYTRUDA, incluse reazioni di Grado 4 (<0,1%), Grado 3 (0,4%) e Grado 2 (0,1%). I corticosteroidi sistemici sono stati richiesti nel 68% (13/19) dei pazienti; ulteriore terapia immunosoppressiva è stata richiesta nell’11% dei pazienti. L’epatite ha portato all’interruzione permanente di KEYTRUDA nello 0,2% (6) e alla sospensione nello 0,3% (9) dei pazienti. Tutti i pazienti che sono stati sospesi hanno ripreso KEYTRUDA dopo il miglioramento dei sintomi; di questi, nessuno ha avuto recidiva. L’epatite si è risolta nel 79% dei 19 pazienti.
KEYTRUDA Con Axitinib KEYTRUDA in combinazione con axitinib può causare tossicità epatica. Monitorare gli enzimi epatici prima dell’inizio e periodicamente durante il trattamento. Considerare il monitoraggio più frequentemente rispetto a quando i farmaci vengono somministrati come singoli agenti. Per enzimi epatici elevati, interrompere KEYTRUDA e axitinib e prendere in considerazione la somministrazione di corticosteroidi secondo necessità. Con la combinazione di KEYTRUDA e axitinib, l’aumento dell’alanina aminotransferasi (ALT) (20%) e l’aumento dell’aspartato aminotransferasi (AST) (13%) di Grado 3 e 4 sono stati osservati con una frequenza maggiore rispetto al solo KEYTRUDA. Il 59% dei pazienti con ALT aumentata ha ricevuto corticosteroidi sistemici. Nei pazienti con ALT ?3 volte il limite superiore della norma (ULN) (Gradi 2-4, n=116), l’ALT si è risolta a Gradi 0-1 nel 94%. Tra i 92 pazienti sottoposti nuovamente a trattamento con KEYTRUDA (n=3) o axitinib (n=34) somministrati come agente singolo o con entrambi (n=55), è stata osservata recidiva di ALT ?3 volte ULN in 1 paziente che riceveva KEYTRUDA , 16 pazienti trattati con axitinib e 24 pazienti trattati con entrambi. Tutti i pazienti con recidiva di ALT ?3 ULN si sono successivamente ripresi dall’evento. Endocrinopatie immuno-mediate Insufficienza surrenalica KEYTRUDA può causare insufficienza surrenalica primaria o secondaria. Per Grado 2 o superiore, iniziare un trattamento sintomatico, inclusa la sostituzione ormonale come clinicamente indicato. Sospendere KEYTRUDA a seconda della gravità. Insufficienza surrenalica si è verificata nello 0,8% (22/2799) dei pazienti trattati con KEYTRUDA, incluse reazioni di Grado 4 (<0,1%), Grado 3 (0,3%) e Grado 2 (0,3%). I corticosteroidi sistemici sono stati richiesti nel 77% (17/22) dei pazienti; di questi, la maggior parte è rimasta in terapia con corticosteroidi sistemici. L’insufficienza surrenalica ha portato all’interruzione permanente di KEYTRUDA in <0,1% (1) e alla sospensione nello 0,3% (8) dei pazienti. Tutti i pazienti che sono stati sospesi hanno ripreso KEYTRUDA dopo il miglioramento dei sintomi.
Ipofisite KEYTRUDA può causare ipofisite immuno-mediata. L’ipofisite può presentarsi con sintomi acuti associati all’effetto massa come mal di testa, fotofobia o difetti del campo visivo. L’ipofisite può causare ipopituitarismo. Avviare la sostituzione ormonale come indicato. Sospendere o sospendere definitivamente KEYTRUDA a seconda della gravità. L’ipofisite si è verificata nello 0,6% (17/2799) dei pazienti trattati con KEYTRUDA, incluse reazioni di Grado 4 (<0,1%), Grado 3 (0,3%) e Grado 2 (0,2%). I corticosteroidi sistemici erano richiesti nel 94% (16/17) dei pazienti; di questi, la maggior parte è rimasta in terapia con corticosteroidi sistemici. L’ipofisite ha portato all’interruzione permanente di KEYTRUDA nello 0,1% (4) e alla sospensione nello 0,3% (7) dei pazienti. Tutti i pazienti che sono stati sospesi hanno ripreso KEYTRUDA dopo il miglioramento dei sintomi.
Disturbi della tiroide KEYTRUDA può causare disturbi della tiroide immuno-mediati. La tiroidite può presentarsi con o senza endocrinopatia. L’ipotiroidismo può seguire l’ipertiroidismo. Iniziare la sostituzione ormonale per l’ipotiroidismo o istituire la gestione medica dell’ipertiroidismo come clinicamente indicato. Sospendere o sospendere definitivamente KEYTRUDA a seconda della gravità. La tiroidite si è verificata nello 0,6% (16/2799) dei pazienti trattati con KEYTRUDA, compreso il Grado 2 (0,3%). Nessuno è stato interrotto, ma KEYTRUDA è stato sospeso in <0,1% (1) dei pazienti.
L’ipertiroidismo si è verificato nel 3,4% (96/2799) dei pazienti trattati con KEYTRUDA, compreso il Grado 3 (0,1%) e il Grado 2 (0,8%). Ha portato all’interruzione permanente di KEYTRUDA in <0,1% (2) e alla sospensione nello 0,3% (7) dei pazienti. Tutti i pazienti che sono stati sospesi hanno ripreso KEYTRUDA dopo il miglioramento dei sintomi. L’ipotiroidismo si è verificato nell’8% (237/2799) dei pazienti trattati con KEYTRUDA, compreso il Grado 3 (0,1%) e il Grado 2 (6,2%). Ha portato all’interruzione permanente di KEYTRUDA in <0,1% (1) e alla sospensione nello 0,5% (14) dei pazienti. Tutti i pazienti che sono stati sospesi hanno ripreso KEYTRUDA dopo il miglioramento dei sintomi. La maggior parte dei pazienti con ipotiroidismo ha richiesto la sostituzione a lungo termine dell’ormone tiroideo. L’incidenza di ipotiroidismo nuovo o in peggioramento era più alta in 1185 pazienti con HNSCC, verificatasi nel 16% dei pazienti trattati con KEYTRUDA in monoterapia o in combinazione con platino e FU, incluso ipotiroidismo di grado 3 (0,3%). L’incidenza di ipotiroidismo nuovo o in peggioramento è stata più alta in 389 pazienti adulti con cHL (17%) trattati con KEYTRUDA in monoterapia, compreso l’ipotiroidismo di grado 1 (6,2%) e di grado 2 (10,8%).
Diabete mellito di tipo 1 (DM), che può presentarsi con chetoacidosi diabetica Monitorare i pazienti per iperglicemia o altri segni e sintomi del diabete. Iniziare il trattamento con insulina come clinicamente indicato. Sospendere KEYTRUDA a seconda della gravità. Il DM di tipo 1 si è verificato nello 0,2% (6/2799) dei pazienti trattati con KEYTRUDA. Ha portato all’interruzione permanente in <0,1% (1) e alla sospensione di KEYTRUDA in <0,1% (1) dei pazienti. Tutti i pazienti che sono stati sospesi hanno ripreso KEYTRUDA dopo il miglioramento dei sintomi.
Nefrite immuno-mediata con disfunzione renale KEYTRUDA può causare nefrite immuno-mediata. Nefrite immuno-mediata si è verificata nello 0,3% (9/2799) dei pazienti trattati con KEYTRUDA, incluse reazioni di Grado 4 (<0,1%), Grado 3 (0,1%) e Grado 2 (0,1%). I corticosteroidi sistemici erano richiesti nell’89% (8/9) dei pazienti. La nefrite ha portato all’interruzione permanente di KEYTRUDA nello 0,1% (3) e alla sospensione nello 0,1% (3) dei pazienti. Tutti i pazienti che sono stati sospesi hanno ripreso KEYTRUDA dopo il miglioramento dei sintomi; di questi, nessuno ha avuto recidiva. La nefrite si è risolta nel 56% dei 9 pazienti.
Reazioni avverse dermatologiche immuno-mediate KEYTRUDA può causare eruzioni cutanee o dermatiti immuno-mediate. Dermatite esfoliativa, inclusa la sindrome di Stevens-Johnson, rash da farmaci con eosinofilia e sintomi sistemici e necrolisi epidermica tossica, si è verificata con i trattamenti anti-PD-1/PD-L1. Emollienti topici e/o corticosteroidi topici possono essere adeguati per il trattamento di rash non esfoliativi da lievi a moderati. Sospendere o sospendere definitivamente KEYTRUDA a seconda della gravità. Reazioni avverse dermatologiche immuno-mediate si sono verificate nell’1,4% (38/2799) dei pazienti trattati con KEYTRUDA, incluse reazioni di grado 3 (1%) e di grado 2 (0,1%). I corticosteroidi sistemici sono stati richiesti nel 40% (15/38) dei pazienti. Queste reazioni hanno portato all’interruzione permanente del trattamento nello 0,1% (2) e alla sospensione di KEYTRUDA nello 0,6% (16) dei pazienti. Tutti i pazienti che sono stati sospesi hanno ripreso KEYTRUDA dopo il miglioramento dei sintomi; di questi, Il 6% ha avuto una recidiva. Le reazioni si sono risolte nel 79% dei 38 pazienti.
Altre reazioni avverse immuno-mediate Le seguenti reazioni avverse immuno-mediate clinicamente significative si sono verificate con un’incidenza <1% (se non diversamente specificato) in pazienti che hanno ricevuto KEYTRUDA o sono state segnalate con l’uso di altri trattamenti anti-PD-1/PD-L1. Per alcune di queste reazioni avverse sono stati segnalati casi gravi o fatali. Cardiaco/Vascolare: Miocardite, pericardite, vasculite; Sistema nervoso: meningite, encefalite, mielite e demielinizzazione, sindrome miastenica/miastenia grave (inclusa esacerbazione), sindrome di Guillain-Barré, paresi nervosa, neuropatia autoimmune; Oculare: possono verificarsi uveite, irite e altre tossicità infiammatorie oculari. Alcuni casi possono essere associati al distacco della retina. Possono verificarsi vari gradi di disabilità visiva, inclusa la cecità. Se l’uveite si verifica in combinazione con altre reazioni avverse immuno-mediate, prendere in considerazione una sindrome simile a Vogt-Koyanagi-Harada, poiché potrebbe richiedere un trattamento con steroidi sistemici per ridurre il rischio di perdita permanente della vista; Gastrointestinale: pancreatite, per includere aumenti dei livelli sierici di amilasi e lipasi, gastrite, duodenite; Tessuto muscoloscheletrico e connettivo: miosite/polimiosite, rabdomiolisi (e sequele associate, inclusa insufficienza renale), artrite (1,5%), polimialgia reumatica; Endocrino: Ipoparatiroidismo; Ematologico/immunitario: anemia emolitica, anemia aplastica, linfoistiocitosi emofagocitica, sindrome da risposta infiammatoria sistemica, linfoadenite necrotizzante istiocitica (linfadenite di Kikuchi), sarcoidosi, porpora trombocitopenica immunitaria, rigetto del trapianto di organo solido. Gastrointestinale: pancreatite, per includere aumenti dei livelli sierici di amilasi e lipasi, gastrite, duodenite; Tessuto muscoloscheletrico e connettivo: miosite/polimiosite, rabdomiolisi (e sequele associate, inclusa insufficienza renale), artrite (1,5%), polimialgia reumatica; Endocrino: Ipoparatiroidismo; Ematologico/immunitario: anemia emolitica, anemia aplastica, linfoistiocitosi emofagocitica, sindrome da risposta infiammatoria sistemica, linfoadenite necrotizzante istiocitica (linfadenite di Kikuchi), sarcoidosi, porpora trombocitopenica immunitaria, rigetto del trapianto di organo solido. Gastrointestinale: pancreatite, per includere aumenti dei livelli sierici di amilasi e lipasi, gastrite, duodenite; Tessuto muscoloscheletrico e connettivo: miosite/polimiosite, rabdomiolisi (e sequele associate, inclusa insufficienza renale), artrite (1,5%), polimialgia reumatica; Endocrino: Ipoparatiroidismo; Ematologico/immunitario: anemia emolitica, anemia aplastica, linfoistiocitosi emofagocitica, sindrome da risposta infiammatoria sistemica, linfoadenite necrotizzante istiocitica (linfadenite di Kikuchi), sarcoidosi, porpora trombocitopenica immunitaria, rigetto del trapianto di organo solido. polimialgia reumatica; Endocrino: Ipoparatiroidismo; Ematologico/immunitario: anemia emolitica, anemia aplastica, linfoistiocitosi emofagocitica, sindrome da risposta infiammatoria sistemica, linfoadenite necrotizzante istiocitica (linfadenite di Kikuchi), sarcoidosi, porpora trombocitopenica immunitaria, rigetto del trapianto di organo solido. polimialgia reumatica; Endocrino: Ipoparatiroidismo; Ematologico/immunitario: anemia emolitica, anemia aplastica, linfoistiocitosi emofagocitica, sindrome da risposta infiammatoria sistemica, linfoadenite necrotizzante istiocitica (linfadenite di Kikuchi), sarcoidosi, porpora trombocitopenica immunitaria, rigetto del trapianto di organo solido.
Reazioni correlate all’infusione KEYTRUDA può causare reazioni correlate all’infusione gravi o pericolose per la vita, incluse ipersensibilità e anafilassi, che sono state riportate nello 0,2% dei 2799 pazienti trattati con KEYTRUDA. Monitorare segni e sintomi di reazioni correlate all’infusione. Interrompere o rallentare la velocità di infusione per reazioni di Grado 1 o Grado 2. Per reazioni di Grado 3 o Grado 4, interrompere l’infusione e sospendere definitivamente KEYTRUDA.
Complicazioni del trapianto allogenico di cellule staminali ematopoietiche (HSCT) Complicanze fatali e altre gravi possono verificarsi in pazienti che ricevono HSCT allogenico prima o dopo i trattamenti anti-PD-1/PD-L1. Le complicanze correlate al trapianto comprendono la malattia iperacuta del trapianto contro l’ospite (GVHD), la GVHD acuta e cronica, la malattia veno-occlusiva epatica dopo condizionamento a intensità ridotta e la sindrome febbrile richiedente steroidi (senza una causa infettiva identificata). Queste complicanze possono verificarsi nonostante la terapia intermedia tra il trattamento anti-PD-1/PD-L1 e l’HSCT allogenico. Seguire attentamente i pazienti per l’evidenza di queste complicanze e intervenire tempestivamente. Considerare i benefici rispetto ai rischi dell’utilizzo di trattamenti anti-PD-1/PD-L1 prima o dopo un trapianto allogenico.
Aumento della mortalità nei pazienti con mieloma multiplo Negli studi condotti su pazienti con mieloma multiplo, l’aggiunta di KEYTRUDA a un analogo del talidomide più desametasone ha determinato un aumento della mortalità. Il trattamento di questi pazienti con un trattamento anti-PD-1/PD-L1 in questa combinazione non è raccomandato al di fuori degli studi controllati.
Tossicità embriofetale Sulla base del suo meccanismo d’azione, KEYTRUDA può causare danni al feto se somministrato a donne in gravidanza. Avvisare le donne di questo potenziale rischio. Nelle donne in età fertile, verificare lo stato di gravidanza prima di iniziare KEYTRUDA e consigliare loro di usare un contraccettivo efficace durante il trattamento e per 4 mesi dopo l’ultima dose.
Reazioni avverse In KEYNOTE-006, KEYTRUDA è stato interrotto a causa di reazioni avverse nel 9% di 555 pazienti con melanoma avanzato; le reazioni avverse che hanno portato all’interruzione permanente in più di un paziente sono state colite (1,4%), epatite autoimmune (0,7%), reazione allergica (0,4%), polineuropatia (0,4%) e insufficienza cardiaca (0,4%). Le reazioni avverse più comuni (?20%) con KEYTRUDA sono state affaticamento (28%), diarrea (26%), eruzione cutanea (24%) e nausea (21%).
Nello studio KEYNOTE-054, quando KEYTRUDA è stato somministrato in monoterapia a pazienti con melanoma in stadio III, KEYTRUDA è stato definitivamente interrotto a causa di reazioni avverse nel 14% di 509 pazienti; i più comuni (?1%) erano polmonite (1,4%), colite (1,2%) e diarrea (1%). Reazioni avverse gravi si sono verificate nel 25% dei pazienti trattati con KEYTRUDA. La reazione avversa più comune (?20%) con KEYTRUDA è stata la diarrea (28%). Nello studio KEYNOTE-716, quando KEYTRUDA è stato somministrato come agente singolo a pazienti con melanoma in stadio IIB o IIC, le reazioni avverse che si sono verificate nei pazienti con melanoma in stadio IIB o IIC sono state simili a quelle che si sono verificate nei 1011 pazienti con melanoma in stadio III dello studio KEYNOTE-054.
Nello studio KEYNOTE-189, quando KEYTRUDA è stato somministrato con pemetrexed e chemioterapia a base di platino nel NSCLC metastatico non squamoso, KEYTRUDA è stato interrotto a causa di reazioni avverse nel 20% dei 405 pazienti. Le reazioni avverse più comuni che hanno portato all’interruzione permanente di KEYTRUDA sono state polmonite (3%) e danno renale acuto (2%). Le reazioni avverse più comuni (?20%) con KEYTRUDA sono state nausea (56%), affaticamento (56%), costipazione (35%), diarrea (31%), diminuzione dell’appetito (28%), eruzione cutanea (25%), vomito (24%), tosse (21%), dispnea (21%) e piressia (20%).
Nello studio KEYNOTE-407, quando KEYTRUDA è stato somministrato con carboplatino e paclitaxel o paclitaxel legato alle proteine ??nel NSCLC squamoso metastatico, KEYTRUDA è stato interrotto a causa di reazioni avverse nel 15% dei 101 pazienti. Le reazioni avverse gravi più frequenti riportate in almeno il 2% dei pazienti sono state neutropenia febbrile, polmonite e infezione del tratto urinario. Le reazioni avverse osservate nello studio KEYNOTE-407 sono state simili a quelle osservate nello studio KEYNOTE-189, con l’eccezione che nel braccio KEYTRUDA e chemioterapia sono state osservate maggiori incidenze di alopecia (47% vs 36%) e neuropatia periferica (31% vs 25%) al braccio placebo e chemioterapia in KEYNOTE-407.
Nello studio KEYNOTE-042, KEYTRUDA è stato interrotto a causa di reazioni avverse nel 19% dei 636 pazienti con NSCLC avanzato; i più comuni sono stati la polmonite (3%), la morte per causa sconosciuta (1,6%) e la polmonite (1,4%). Le reazioni avverse gravi più frequenti riportate in almeno il 2% dei pazienti sono state polmonite (7%), polmonite (3,9%), embolia polmonare (2,4%) e versamento pleurico (2,2%). La reazione avversa più comune (?20%) è stata l’affaticamento (25%).
Nello studio KEYNOTE-010, la monoterapia con KEYTRUDA è stata interrotta a causa di reazioni avverse nell’8% dei 682 pazienti con NSCLC metastatico; la più comune era la polmonite (1,8%). Le reazioni avverse più comuni (?20%) sono state diminuzione dell’appetito (25%), affaticamento (25%), dispnea (23%) e nausea (20%).
Nello studio KEYNOTE-048, la monoterapia con KEYTRUDA è stata interrotta a causa di eventi avversi nel 12% dei 300 pazienti con HNSCC; le reazioni avverse più comuni che hanno portato all’interruzione definitiva sono state sepsi (1,7%) e polmonite (1,3%). Le reazioni avverse più comuni (?20%) sono state affaticamento (33%), costipazione (20%) ed eruzione cutanea (20%).
Nello studio KEYNOTE-048, quando KEYTRUDA è stato somministrato in combinazione con platino (cisplatino o carboplatino) e chemioterapia con FU, KEYTRUDA è stato interrotto a causa di reazioni avverse nel 16% dei 276 pazienti con HNSCC. Le reazioni avverse più comuni che hanno portato all’interruzione permanente di KEYTRUDA sono state polmonite (2,5%), polmonite (1,8%) e shock settico (1,4%). Le reazioni avverse più comuni (?20%) sono state nausea (51%), affaticamento (49%), costipazione (37%), vomito (32%), infiammazione della mucosa (31%), diarrea (29%), diminuzione dell’appetito (29%), stomatite (26%) e tosse (22%).
Nello studio KEYNOTE-012, KEYTRUDA è stato interrotto a causa di reazioni avverse nel 17% dei 192 pazienti con HNSCC. Reazioni avverse gravi si sono verificate nel 45% dei pazienti. Le reazioni avverse gravi più frequenti riportate in almeno il 2% dei pazienti sono state polmonite, dispnea, stato confusionale, vomito, versamento pleurico e insufficienza respiratoria. Le reazioni avverse più comuni (?20%) sono state affaticamento, diminuzione dell’appetito e dispnea. Le reazioni avverse che si sono verificate nei pazienti con HNSCC sono state generalmente simili a quelle che si sono verificate nei pazienti con melanoma o NSCLC che hanno ricevuto KEYTRUDA in monoterapia, ad eccezione dell’aumentata incidenza di edema facciale e ipotiroidismo nuovo o in peggioramento.
Nello studio KEYNOTE-204, KEYTRUDA è stato interrotto a causa di reazioni avverse nel 14% dei 148 pazienti con cHL. Reazioni avverse gravi si sono verificate nel 30% dei pazienti trattati con KEYTRUDA; quelli ?1% erano polmonite, polmonite, piressia, miocardite, danno renale acuto, neutropenia febbrile e sepsi. Tre pazienti sono deceduti per cause diverse dalla progressione della malattia: 2 per complicazioni dopo trapianto allogenico e 1 per causa sconosciuta. Le reazioni avverse più comuni (?20%) sono state infezione del tratto respiratorio superiore (41%), dolore muscoloscheletrico (32%), diarrea (22%) e piressia, affaticamento, eruzione cutanea e tosse (20% ciascuna).
Nello studio KEYNOTE-087, KEYTRUDA è stato interrotto a causa di reazioni avverse nel 5% dei 210 pazienti con cHL. Reazioni avverse gravi si sono verificate nel 16% dei pazienti; quelli ?1% erano polmonite, polmonite, piressia, dispnea, GVHD e herpes zoster. Due pazienti sono deceduti per cause diverse dalla progressione della malattia: 1 per GVHD dopo successivo HSCT allogenico e 1 per shock settico. Le reazioni avverse più comuni (?20%) sono state affaticamento (26%), piressia (24%), tosse (24%), dolore muscoloscheletrico (21%), diarrea (20%) ed eruzione cutanea (20%).
Nello studio KEYNOTE-170, KEYTRUDA è stato interrotto a causa di reazioni avverse nell’8% dei 53 pazienti con PMBCL. Reazioni avverse gravi si sono verificate nel 26% dei pazienti e comprendevano aritmia (4%), tamponamento cardiaco (2%), infarto del miocardio (2%), versamento pericardico (2%) e pericardite (2%). Sei (11%) pazienti sono deceduti entro 30 giorni dall’inizio del trattamento. Le reazioni avverse più comuni (?20%) sono state dolore muscoloscheletrico (30%), infezione del tratto respiratorio superiore e piressia (28% ciascuno), tosse (26%), affaticamento (23%) e dispnea (21%).
Nello studio KEYNOTE-052, KEYTRUDA è stato interrotto a causa di reazioni avverse nell’11% di 370 pazienti con malattia localmente avanzata o mUC. Reazioni avverse gravi si sono verificate nel 42% dei pazienti; quelli ?2% erano infezione del tratto urinario, ematuria, danno renale acuto, polmonite e urosepsi. Le reazioni avverse più comuni (?20%) sono state affaticamento (38%), dolore muscoloscheletrico (24%), diminuzione dell’appetito (22%), costipazione (21%), eruzione cutanea (21%) e diarrea (20%).
Nello studio KEYNOTE-045, KEYTRUDA è stato interrotto a causa di reazioni avverse nell’8% dei 266 pazienti con malattia localmente avanzata o mUC. La reazione avversa più comune che ha portato all’interruzione permanente di KEYTRUDA è stata la polmonite (1,9%). Reazioni avverse gravi si sono verificate nel 39% dei pazienti trattati con KEYTRUDA; quelli ?2% erano infezioni del tratto urinario, polmonite, anemia e polmonite. Le reazioni avverse più comuni (?20%) nei pazienti che hanno ricevuto KEYTRUDA sono state affaticamento (38%), dolore muscoloscheletrico (32%), prurito (23%), diminuzione dell’appetito (21%), nausea (21%) e rash (20%).
Nello studio KEYNOTE-057, KEYTRUDA è stato interrotto a causa di reazioni avverse nell’11% dei 148 pazienti con NMIBC ad alto rischio. La reazione avversa più comune che ha portato all’interruzione permanente di KEYTRUDA è stata la polmonite (1,4%). Reazioni avverse gravi si sono verificate nel 28% dei pazienti; quelli ?2% erano polmonite (3%), ischemia cardiaca (2%), colite (2%), embolia polmonare (2%), sepsi (2%) e infezione del tratto urinario (2%). Le reazioni avverse più comuni (?20%) sono state affaticamento (29%), diarrea (24%) ed eruzione cutanea (24%).
Le reazioni avverse verificatesi in pazienti con MSI-H o dMMR CRC sono state simili a quelle verificatesi in pazienti con melanoma o NSCLC che hanno ricevuto KEYTRUDA in monoterapia.
Nello studio KEYNOTE-811, quando KEYTRUDA è stato somministrato in combinazione con trastuzumab, chemioterapia contenente fluoropirimidina e platino, KEYTRUDA è stato interrotto a causa di reazioni avverse nel 6% di 217 pazienti con adenocarcinoma HER2+ gastrico o GEJ localmente avanzato non resecabile o metastatico. La reazione avversa più comune che ha portato all’interruzione permanente è stata la polmonite (1,4%). Nel braccio KEYTRUDA rispetto al placebo, è stata osservata una differenza di incidenza ?5% tra i pazienti trattati con KEYTRUDA rispetto allo standard di cura per diarrea (53% vs 44%) e nausea (49% vs 44%).
Le reazioni avverse più comuni (riportate in ?20%) nei pazienti trattati con KEYTRUDA in combinazione con chemioterapia sono state affaticamento/astenia, nausea, costipazione, diarrea, diminuzione dell’appetito, eruzione cutanea, vomito, tosse, dispnea, piressia, alopecia, neuropatia periferica, infiammazione, stomatite, mal di testa, perdita di peso, dolore addominale, artralgia, mialgia e insonnia.
Nello studio KEYNOTE-590, quando KEYTRUDA è stato somministrato con cisplatino e fluorouracile a pazienti con carcinoma esofageo metastatico o localmente avanzato o GEJ (tumori con epicentro da 1 a 5 centimetri al di sopra del GEJ) che non erano candidati alla resezione chirurgica o alla chemioradioterapia definitiva, KEYTRUDA è stato interrotto a causa di reazioni avverse nel 15% di 370 pazienti. Le reazioni avverse più comuni che hanno portato all’interruzione permanente di KEYTRUDA (?1%) sono state polmonite (1,6%), danno renale acuto (1,1%) e polmonite (1,1%). Le reazioni avverse più comuni (?20%) con KEYTRUDA in combinazione con chemioterapia sono state nausea (67%), affaticamento (57%), diminuzione dell’appetito (44%), costipazione (40%), diarrea (36%), vomito ( 34%), stomatite (27%) e perdita di peso (24%).
Le reazioni avverse che si sono verificate in pazienti con carcinoma esofageo che hanno ricevuto KEYTRUDA in monoterapia sono state simili a quelle verificatesi in pazienti con melanoma o NSCLC che hanno ricevuto KEYTRUDA in monoterapia.
Reazioni avverse gravi si sono verificate nel 50% dei pazienti trattati con KEYTRUDA in combinazione con chemioterapia con o senza bevacizumab; quelli ?3% erano neutropenia febbrile (6,8%), infezione del tratto urinario (5,2%), anemia (4,6%) e danno renale acuto e sepsi (3,3% ciascuno).
KEYTRUDA è stato interrotto nel 15% dei pazienti a causa di reazioni avverse. La reazione avversa più comune che ha portato all’interruzione permanente (?1%) è stata la colite (1%).
Per i pazienti trattati con KEYTRUDA, chemioterapia e bevacizumab (n=196), le reazioni avverse più comuni (?20%) sono state neuropatia periferica (62%), alopecia (58%), anemia (55%), affaticamento/astenia ( 53%), nausea e neutropenia (41% ciascuno), diarrea (39%), ipertensione e trombocitopenia (35% ciascuno), costipazione e artralgia (31% ciascuno), vomito (30%), infezione delle vie urinarie (27%) , rash (26%), leucopenia (24%), ipotiroidismo (22%) e diminuzione dell’appetito (21%).
Per i pazienti trattati con KEYTRUDA in combinazione con chemioterapia con o senza bevacizumab, le reazioni avverse più comuni (?20%) sono state neuropatia periferica (58%), alopecia (56%), affaticamento (47%), nausea (40%), diarrea (36%), costipazione (28%), artralgia (27%), vomito (26%), ipertensione e infezione del tratto urinario (24% ciascuna) ed eruzione cutanea (22%).
Nello studio KEYNOTE-158, KEYTRUDA è stato interrotto a causa di reazioni avverse nell’8% di 98 pazienti con carcinoma cervicale ricorrente o metastatico precedentemente trattato. Reazioni avverse gravi si sono verificate nel 39% dei pazienti trattati con KEYTRUDA; i più frequenti includevano anemia (7%), fistole, emorragie e infezioni [tranne le infezioni del tratto urinario] (4,1% ciascuno). Le reazioni avverse più comuni (?20%) sono state affaticamento (43%), dolore muscoloscheletrico (27%), diarrea (23%), dolore e dolore addominale (22% ciascuno) e diminuzione dell’appetito (21%).
Le reazioni avverse che si sono verificate nei pazienti con HCC sono state generalmente simili a quelle nei pazienti con melanoma o NSCLC che hanno ricevuto KEYTRUDA in monoterapia, con l’eccezione dell’aumentata incidenza di ascite (8% Gradi 3-4) ed epatite immuno-mediata (2,9%) . Le anomalie di laboratorio (Gradi 3-4) che si sono verificate con una maggiore incidenza sono state elevate AST (20%), ALT (9%) e iperbilirubinemia (10%).
Tra i 50 pazienti con MCC arruolati nello studio KEYNOTE-017, le reazioni avverse che si sono verificate nei pazienti con MCC sono state generalmente simili a quelle che si sono verificate nei pazienti con melanoma o NSCLC che hanno ricevuto KEYTRUDA in monoterapia. Le anomalie di laboratorio (Gradi 3-4) che si sono verificate con una maggiore incidenza sono state AST elevata (11%) e iperglicemia (19%).
Nello studio KEYNOTE-426, quando KEYTRUDA è stato somministrato in combinazione con axitinib, si sono verificate reazioni avverse fatali nel 3,3% di 429 pazienti. Reazioni avverse gravi si sono verificate nel 40% dei pazienti, le più frequenti (?1%) sono state epatotossicità (7%), diarrea (4,2%), danno renale acuto (2,3%), disidratazione (1%) e polmonite (1% ). L’interruzione permanente a causa di una reazione avversa si è verificata nel 31% dei pazienti; solo KEYTRUDA (13%), solo axitinib (13%) e la combinazione (8%); i più comuni erano epatotossicità (13%), diarrea/colite (1,9%), danno renale acuto (1,6%) e accidente cerebrovascolare (1,2%). Le reazioni avverse più comuni (?20%) sono state diarrea (56%), affaticamento/astenia (52%), ipertensione (48%), epatotossicità (39%), ipotiroidismo (35%), diminuzione dell’appetito (30%), eritrodisestesia palmo-plantare (28%), nausea (28%),
Nello studio KEYNOTE-564, quando KEYTRUDA è stato somministrato come agente singolo per il trattamento adiuvante del carcinoma a cellule renali, si sono verificate reazioni avverse gravi nel 20% dei pazienti trattati con KEYTRUDA; le reazioni avverse gravi (?1%) sono state danno renale acuto, insufficienza surrenalica, polmonite, colite e chetoacidosi diabetica (1% ciascuna). Reazioni avverse fatali si sono verificate nello 0,2% incluso 1 caso di polmonite. L’interruzione di KEYTRUDA a causa di reazioni avverse si è verificata nel 21% di 488 pazienti; i più comuni (?1%) sono stati l’aumento di ALT (1,6%), la colite (1%) e l’insufficienza surrenalica (1%). Le reazioni avverse più comuni (?20%) sono state dolore muscoloscheletrico (41%), affaticamento (40%), eruzione cutanea (30%), diarrea (27%), prurito (23%) e ipotiroidismo (21%).
Le reazioni avverse che si sono verificate in pazienti con carcinoma endometriale MSI-H o dMMR che hanno ricevuto KEYTRUDA come agente singolo sono state simili a quelle che si sono verificate in pazienti con melanoma o NSCLC che hanno ricevuto KEYTRUDA come agente singolo.
Le reazioni avverse che si sono verificate in pazienti con tumore TMB-H sono state simili a quelle verificatesi in pazienti con altri tumori solidi che hanno ricevuto KEYTRUDA in monoterapia.
Le reazioni avverse che si sono verificate in pazienti con cSCC ricorrente o metastatico o con cSCC localmente avanzato sono state simili a quelle verificatesi in pazienti con melanoma o NSCLC che hanno ricevuto KEYTRUDA in monoterapia.
Nello studio KEYNOTE-522, quando KEYTRUDA è stato somministrato con chemioterapia neoadiuvante (carboplatino e paclitaxel seguiti da doxorubicina o epirubicina e ciclofosfamide) seguita da intervento chirurgico e trattamento adiuvante continuato con KEYTRUDA come agente singolo (n=778) a pazienti con nuova diagnosi, non trattati in precedenza , TNBC in stadio iniziale ad alto rischio, reazioni avverse fatali si sono verificate nello 0,9% dei pazienti, tra cui 1 ciascuna di crisi surrenalica, encefalite autoimmune, epatite, polmonite, polmonite, embolia polmonare e sepsi in associazione con sindrome da disfunzione multiorgano e infarto del miocardio . Reazioni avverse gravi si sono verificate nel 44% dei pazienti trattati con KEYTRUDA; quelli ?2% erano neutropenia febbrile (15%), piressia (3,7%), anemia (2,6%) e neutropenia (2,2%). KEYTRUDA è stato interrotto nel 20% dei pazienti a causa di reazioni avverse. Le reazioni più comuni (?1%) che hanno portato all’interruzione permanente sono state aumento di ALT (2,7%), aumento di AST (1,5%) ed eruzione cutanea (1%). Le reazioni avverse più comuni (?20%) nei pazienti trattati con KEYTRUDA sono state affaticamento (70%), nausea (67%), alopecia (61%), eruzione cutanea (52%), costipazione (42%), diarrea e neuropatia periferica ( 41% ciascuno), stomatite (34%), vomito (31%), mal di testa (30%), artralgia (29%), piressia (28%), tosse (26%), dolore addominale (24%), diminuzione dell’appetito (23%), insonnia (21%) e mialgia (20%).
Nello studio KEYNOTE-355, quando KEYTRUDA e chemioterapia (paclitaxel, paclitaxel legato alle proteine ??o gemcitabina e carboplatino) sono stati somministrati a pazienti con TNBC non resecabile o metastatico ricorrente localmente che non erano stati precedentemente trattati con chemioterapia nel contesto metastatico (n=596) , reazioni avverse fatali si sono verificate nel 2,5% dei pazienti, inclusi arresto cardiorespiratorio (0,7%) e shock settico (0,3%). Reazioni avverse gravi si sono verificate nel 30% dei pazienti trattati con KEYTRUDA in combinazione con chemioterapia; le reazioni gravi in ???2% sono state polmonite (2,9%), anemia (2,2%) e trombocitopenia (2%). KEYTRUDA è stato interrotto nell’11% dei pazienti a causa di reazioni avverse. Le reazioni più comuni che hanno portato all’interruzione permanente (?1%) sono state aumento di ALT (2,2%), aumento di AST (1,5%) e polmonite (1,2%).
Allattamento A causa della possibilità di gravi reazioni avverse nei bambini allattati al seno, consigliare alle donne di non allattare durante il trattamento e per 4 mesi dopo la dose finale.
Uso pediatrico Nello studio KEYNOTE-051, 161 pazienti pediatrici (62 pazienti pediatrici di età compresa tra 6 mesi e meno di 12 anni e 99 pazienti pediatrici di età compresa tra 12 e 17 anni) hanno ricevuto KEYTRUDA 2 mg/kg ogni 3 settimane. La durata mediana dell’esposizione è stata di 2,1 mesi (intervallo: da 1 giorno a 24 mesi).
Le reazioni avverse che si sono verificate con una frequenza ?10% più alta nei pazienti pediatrici rispetto agli adulti sono state piressia (33%), vomito (30%), leucopenia (30%), infezione del tratto respiratorio superiore (29%), neutropenia (26% ), mal di testa (25%) e anemia di grado 3 (17%).
L’Agenzia di Stampa Parlamentare Agenparl è una delle voci storiche ed autorevoli dell’informazione italiana parlamentare ed è una delle principali news company italiane. Nel 1950 Francesco Lisi fondò la più antica Agenzia giornalistica parlamentare italiana, con il nome di S.P.E.; con l’ingresso nell’ASP (Associazione stampa parlamentare) nel 1953 ne mutò il nome in Agenparl.
Dal 1955 affianca con i suoi notiziari il mondo istituzionale, editoriale, economico e finanziario, diventando oggi una tra le fonti più autorevoli dell’informazione con i propri prodotti, servizi e soluzioni all’avanguardia. Dal 2009 il Direttore è Luigi Camilloni che ha proseguito lungo la strada tracciata da Lisi e cioè quella che da sempre ha contraddistinto l’Agenzia, ossia l’imparzialità.
Una formula editoriale veloce ed innovativa che garantisce un’informazione puntuale e degli approfondimenti originali. Per noi di Agenparl, fare informazione significa mantenere un alto livello di esattezza, obiettività e imparzialità, attraverso un codice linguistico chiaro, ma soprattutto senza far ricorso a formule e luoghi comuni giornalistici.
Agenparl dispone di contenuti, servizi e strumenti a cui si affidano Enti, Istituzioni ed Università, sviluppando una serie di soluzioni personalizzabili a seconda delle necessità dei clienti.
 (AGENPARL) - Roma, 5 Giugno 2023
(AGENPARL) - Roma, 5 Giugno 2023